Information activities
资讯活动



发表文章:
Analyzing the Li−Al−O Interphase of Atomic Layer-Deposited Al2O3Films on Layered Oxide Cathodes Using Atomistic Simulations
发表期刊:
ACS Applied Materials & Interfaces
原文链接:
https://doi.org/10.1021/acsami.3c15080
核心问题: 为什么超薄(<2 nm)的原子层沉积(ALD)Al2O3 涂层能改善锂离子电池层状氧化物阴极的电化学性能?其原子尺度界面结构是什么?
研究方法:采用分子动力学(MD)模拟作为核心研究手段,以克服实验上表征超薄(~1 nm)薄膜界面的困难。使用 INTERFACE 力场(IFF),并验证其对于 LiCoO2 和 Al2O3 体系结构、能量属性的可靠性。构建不同厚度(1-10个单层)的非晶 Al2O3 薄膜与 LiCoO2 (LCO)的(0001)表面接触的模型体系。设计一套退火与平衡化模拟流程,以生成能量稳定的非晶 Li-Al-O 薄膜结构。
一、背景介绍
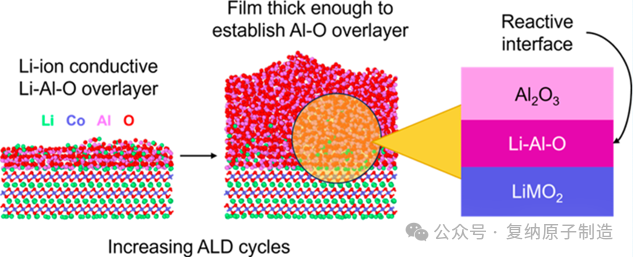
原子层沉积(ALD)的超薄氧化铝 ALD 薄膜(<2 nm)被证明可以提高LiNixMnyCo1-x−yO2 材料的电化学性能,然而,这些薄膜的原子结构尚不清楚,量化超薄(约 1 nm)ALD 薄膜的界面是一项艰巨的实验任务。
传统观点认为,湿化学法制备的涂层需经退火形成 Li-Al-O 界面相以促进锂离子传导,而 ALD 涂层在未退火时同样有效,这引发了对其界面原子结构的深入探究。有研究(如 Hoskins et al.)通过实验发现,在 ALD 过程中,锂存在于薄膜表面,而过渡金属被完全覆盖,这暗示了非纯 Al2O3,可能是 Li-Al-O 化合物的形成。但关于这种超薄 ALD 薄膜的精确原子结构、化学计量比(Li/Al 比)、以及其如何影响锂离子传输,仍然缺乏清晰的理解。
因此,本研究旨在通过原子尺度模拟,揭示 ALD Al2O3 涂层与正极材料间的界面纳米结构,从机理层面解释其性能提升的原因,填补实验观察与理论认知之间的关键空白。
二、研究重点
科罗拉多大学博尔德分校 Alan W. Weimer 团队采用分子动力学对 LiCoO2(LCO)表面接触的不同厚度非晶氧化铝薄膜进行模拟,以量化薄膜纳米结构。
计算了薄膜中的元素质量密度分布,观察到在阴极材料表面,ALD 过程并非机械地包覆氧化铝原子层。相反,来自阴极的 Li 原子会主动“融入”正在生长的氧化铝薄膜中,在 LCO 表面延伸约 2 nm 形成一种有益的 Li-Al-O 界面,并且 Al 和 O 原子在 LCO 薄膜界面处分层,延伸约 1.5 nm。
在这里,原子排列并非完全无序,而是表现出伪晶结构,近似于下面的层状 LiCoO2 晶格,但并不完全相同。更重要的是,锂离子在这个区域中具有异常高的迁移能力。
此外,在足够的厚度下,Li−Al-O 薄膜会转变为非晶氧化铝结构。由于与LiCoO2 具有共享的晶体结构,因此,类似ALD的Li−Al−O薄膜纳米结构的研究也可以应用于其他层状 LiNixMnyCo1−x−yO2 材料。这项工作提供了对非晶 ALD 氧化铝薄膜纳米结构的深入研究,以帮助了解它们作为锂离子电池阴极活性材料保护涂层的用途。
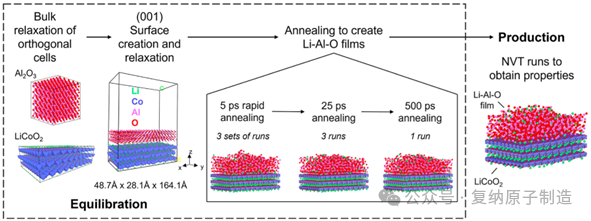
图1. 用于生成与(0001)LiCoO2 表面接触的非晶 Li−Al−O 薄膜的仿真工作流程。为简单起见,模拟单元被用作具有等效 (001) 表面的正交超级单元。Li 原子被着色为绿色,Co 原子被着色为蓝色,Al 原子被着色为粉红色,O 原子被着色为红色。

图2.(a−g)与(001)LCO 表面接触的氧化铝量从 1−6 到 10 个 Al2O3 单层变化。(h)在整个 1000 帧生产模拟中,每 100 帧计算一次模拟非晶氧化铝膜的密度。
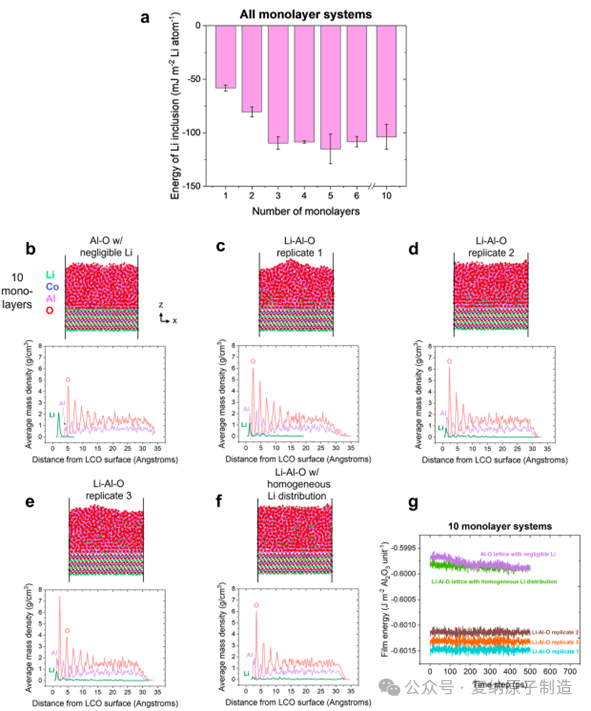
图3.(a)计算非晶 Al−O 晶格中包含锂的能量 E。(b−f)研究了最低能量 10 单层结构,并提供了每个轨迹的快照。(g)绘制了每个轨迹最后 500 ps 的薄膜能量。
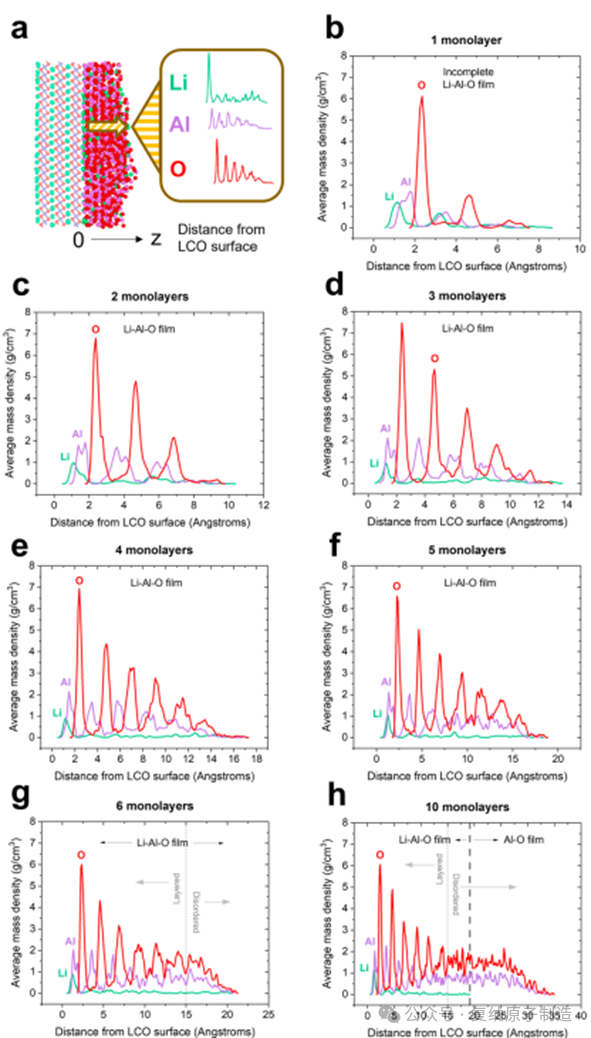
图4. 沿远离 LiCoO2 表面的 z 方向计算的薄膜元素质量密度分布。(a)图形表示如何通过 Li−Al−O 薄膜计算元素质量密度。(b−h)质量密度分布在整个 1000 帧轨迹上进行平均。(g,h)这种 Al 和 O 层在转变为无序结构之前延伸约 1.5 nm。(h)Li-Al-O 区域从 LCO 表面延伸约 2 nm,然后过渡到薄膜表面的无锂 Al-O 区域。
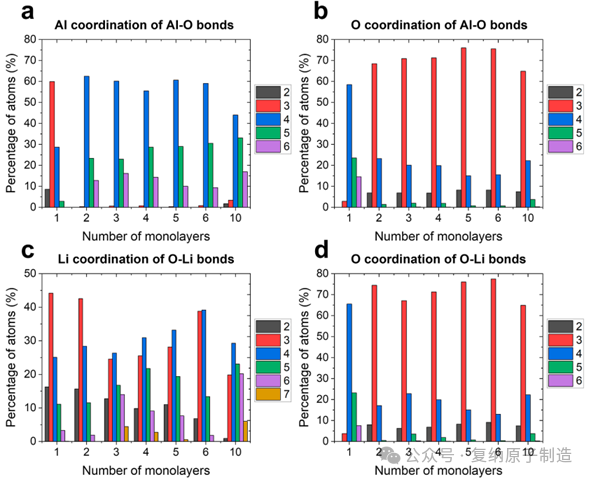
图5.(a)Al−O键的 Al 配位数分布。 Al 最常见的配位环境是[4]Al,其次是[5]Al,然后是[6]Al。 (b)Al−O 键的 O 配位数。 [3]O 是主要的协调环境。(c)O−Li 键的 Li 配位数。 配位数的大分布表明 Li 在 Li−Al−O 晶格中采用了多种构型。 (d)O−Li 键的 O 配位数与 Al−O 键的 O 配位数相似。
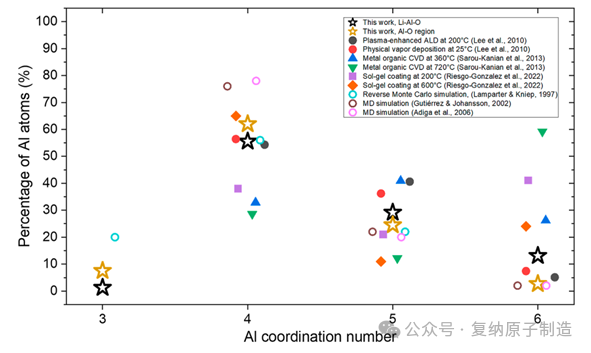
图6. 实验测量和模拟了非晶态氧化铝薄膜中的铝配位数。
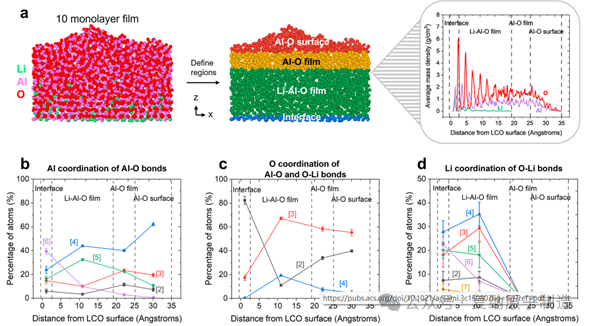
图7. 协调环境作为穿过模拟 10 单层薄膜的距离的函数。 (a)根据其质量密度分布,薄膜被分成四个区域(四个区域的颜色被选择来突出显示不同的区域,并且不对应于配位数)。(b)Al-O 键中 Al 与 O 原子的配位数分布,作为距 LCO 表面距离的函数。(c)O 配位原子的数量与 Al-O 和 O-Li 键的 Al 和 Li 原子之和的分布,作为距 LCO 表面距离的函数。(d)O−Li 键的 Li 配位数随距表面距离的变化的分布。
三、结论
综上所述,本研究通过分子动力学模拟,首次在原子层面证实:在原子层沉积(ALD)过程中,锂(Li)会从层状氧化物阴极(LiCoO2)体相扩散进入生长的非晶Al2O3 薄膜,形成一个厚度约 2 nm 的 Li-Al-O 混合界面区。该区域并非均匀的纯Al2O3 或已知的晶体 LiAlO2,而是一种独特的“伪晶”非晶结构。
独特的“伪晶”结构是一个主动响应界面,通过弹性应变匹配基底、调控局部化学环境、形成梯度功能相,从而能够协同抑制副反应、促进离子传输、增强机械与化学稳定性。这也证明为何超薄 ALD Al2O3 涂层能取得最佳效果——因为在这个尺度下,涂层整体处于具有有益特性的伪晶态,过厚则会弛豫为性能不同的体相结构。
在靠近阴极的界面约 1.5 nm 内,Al 和 O原子受底层晶格“模板”影响,呈现层状排列趋势。但整体上,该区域以四配位 Al 为主导,与块体非晶 Al2O3 结构特征一致。六配位 Al 主要富集在薄膜-阴极的直接界面处,向薄膜表面逐渐减少。锂离子在该界面区内具有宽泛的配位环境和较高的迁移性。当 ALD 涂层足够厚时(如模拟的 10 个单层,约 3 nm),会形成 “梯度双层结构”:靠近阴极的 Li-Al-O 区逐渐过渡到靠近电解质的、几乎无锂的纯非晶 Al-O 区。
计算表明,将锂嵌入 Al-O 网络在热力学上是有利的(负的锂嵌入能),且锂梯度分布的结构比锂均匀分布或完全无锂的结构能量更低、更稳定。晶体α-Al2O3 与 LiCoO2 基底之间存在巨大的晶格失配应变(~69%),而 α-LiAlO2 与基底几乎匹配。Li-Al-O 界面相的形成,是一种能量驱动的自适应过程,能有效缓解界面应变,从而稳定整个涂层体系。
该原子模型完美解释了ALD Al2O3 涂层为何能同时实现 “保护”与“导通” 的双重功能,以及其著名的 “厚度-性能”优化关系:外层(或薄涂层整体)的 Al-O 网络覆盖了阴极表面高活性的过渡金属位点,抑制了电解液副反应。 内层的 Li-Al-O 界面区具有比纯 Al2O3 更高的锂离子电导率,为锂离子在阴极与电解质之间的传输提供了快速通道。 超薄涂层(<2 nm) 整体处于高离子导通的 Li-Al-O 区,故效果最佳。涂层过厚时,表面形成的纯 Al-O 区会增加锂离子传输阻力,导致收益递减。这解释了为何低 ALD 循环次数(2-6次)的涂层效果最为显著。
为“ALD 超薄 Al2O3 涂层改善电池性能”这一长期观察到的实验现象,提供了统一、定量的原子尺度机理模型。由于 LiCoO2 与商用 NMC 材料具有相同的晶体结构,该界面模型预计可广泛适用于层状氧化物正极体系。研究明确指出,最优涂层旨在促进有益 Li-Al-O 界面相的形成,而非单纯沉积更厚的 Al2O3。这为未来设计更高效、更经济的电池表面工程策略(如涂层材料选择、工艺优化)提供了关键的理论基础和设计原则。
了解更多原子层沉积技术以及 Forge Nano 产品详情、应用案例与代包覆服务,欢迎扫描下方二维码填写信息。


您想了解更多信息
请关注我们公众号

如果您想要了解更多产品信息,请填写以下信息下载产品手册, 我们收到您的信息后将第一时间回复您。